Antisense Oligonucleotides (ASO)

Abstract
Antisense oligonucleotides (ASOs) have been validated as therapeutic agents and an important tool in molecular biology. Indeed, ASOs are used either in vitro or in vivo to generate mRNA selective knockouts. They can be used for human therapy since ASOs can inhibit specifically target genes especially whose are difficult to target with small molecules inhibitors or neutralizing antibodies. However, despite their specificity and broadness of use, some practical obstacles remain unsolved in antisense pharmacology, such as insufficient stability due to nucleases degradation activity, and poor cellular delivery as a result of low cellular uptake difficult biological membrane crossing. Moreover, in many cases, potential off-target effects and immunostimulation are also part of the problems derived from their use. In this review, we will discuss ASOs, their chemistry, limitation of use, some solutions to increase stability, and finally some of their therapeutical application.
Keywords
- antisense oligonucleotide
- ASO chemistry
- ASO delivery
- therapeutic target
- cancer
Chapter and author infoShow +
1. Introduction
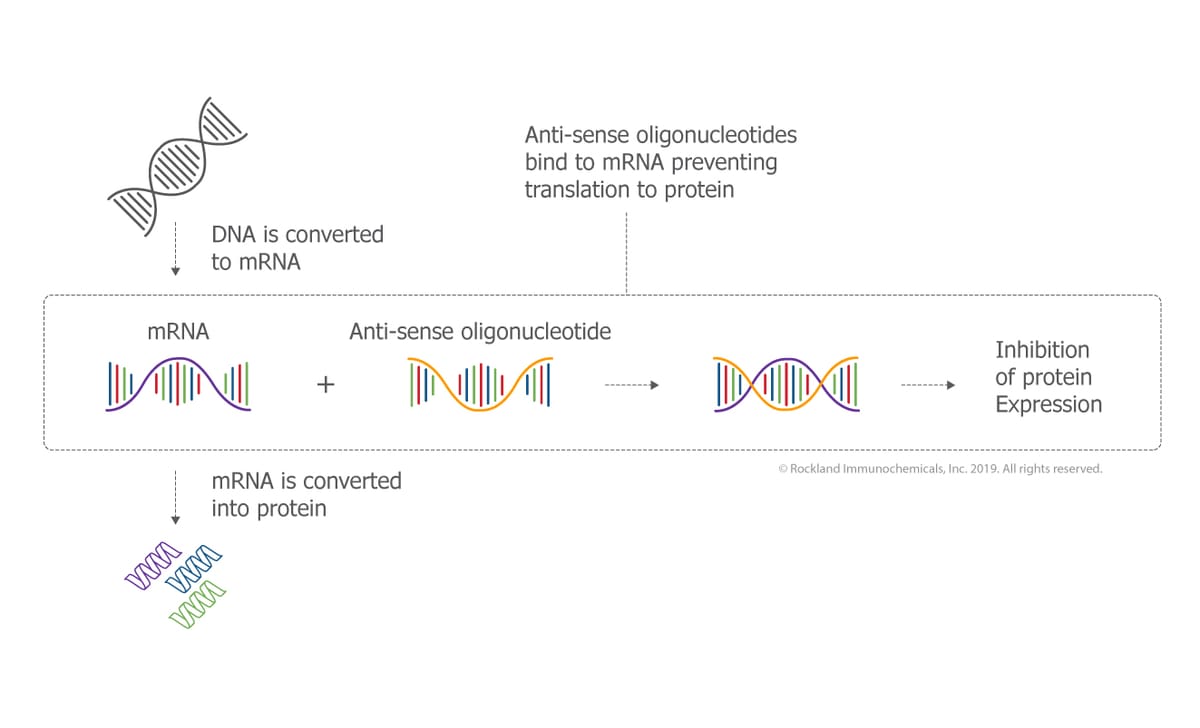
The usage of nucleic acids in therapeutics is founded on the inhibition of RNA expression and on their capacity to modulate the expression of a target protein associate to a disease [1]. ASOs are typically modified synthetic nucleic acids. They are single stand oligonucleotide with 16-mer to 21-mer sequence with high affinity for a specific RNA target sequence, through base pairing [2]. The two general chemical classes of nucleic acids commonly used today are antisense oligonucleotides (ASOs) and siRNA. The first is single-stranded, and modulates RNA function either by degrading the RNA sequence in question due to enzyme RNase H activity, or by modifying splicing function thus affecting RNA metabolism; and the second is double-stranded synthetic oligonucleotides that degrade target RNA through an RNA-induced silencing complex (RISC). Importantly, both must reach the nucleus and/or cytoplasm to exert their activity, and thus must cross a biological membrane.
Although ASOs and siRNA share similarities, they are divergent on some points, and so, choosing ASO or siRNA strategy for gene targeting depends on the target gene. However, since ASOs are single stranded, as opposed to siRNA, they have lower cost of production. Moreover, it is easier to deliver ASOs in vivo, since they do not a vector and a simple chemical modification can increase their resistance to nucleases, as opposed to siRNA that need a carrier. Finally, for in vitro studies, siRNA is considered a better technology, since it’s relatively easier to obtain a potent siRNA since unmodified RNA works with high potency as opposed to ASOs.
In this review we will mainly focus on ASOs chemistry and mechanism of action.
Indeed, since Zamecnik in 1978 used an ASO-like unmodified DNA sequence in cell culture, notable progress has been made in ASO pharmacology [3]. Currently, the efficacy of different ASOs is being studied in many neurodegenerative diseases such as Huntington’s disease, Parkinson’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis but also in several cancer states. Numerous ASO-based therapeutics are being tested in clinical trials. In Table 1 figures some of the ASO in clinical trial for cancer treatments. However, until now, only two ASOs have been approved by the US FDA to be used on humans, namely, fomivirsen (Vitravene), a first-generation ASO for cytomegalovirus (CMV) retinitis, and mipomersen (Kynamro), a second-generation ASO for homozygous familial hypercholesterolemia (HoFH) [4, 5], both developed by Isis Pharmaceuticals. They work via RNase H-mediated cleavage of the targeted RNA.
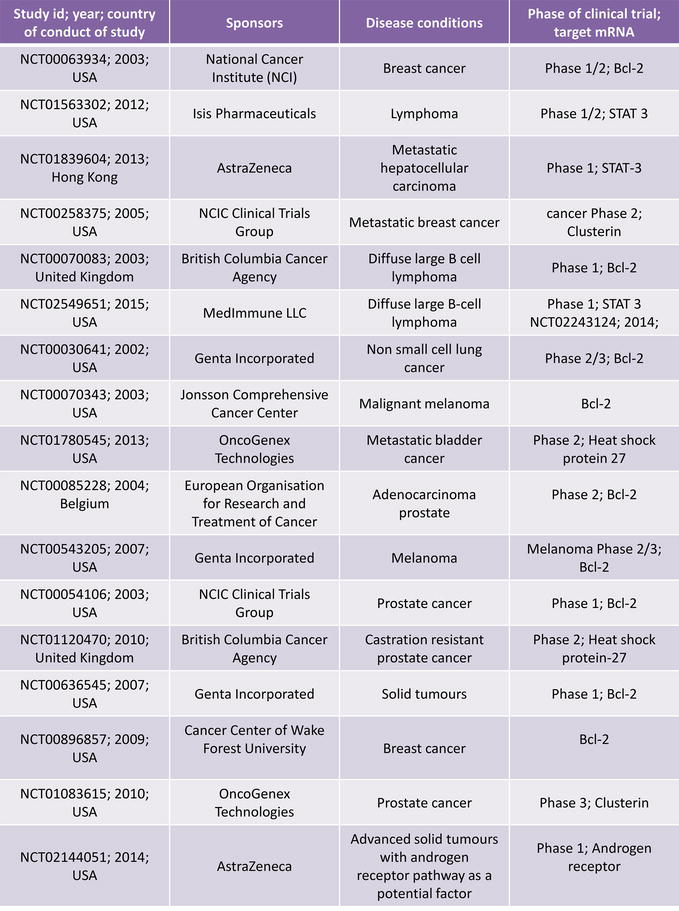
Table 1.
Registered clinical studies with ASO in cancer treatments in clinical trials.gov.
2. ASO’s design, chemistry and mechanism of action
2.1. ASO’s mechanism of action
Two major mechanisms contribute to the antisense activity. The first is that most ASOs are designed to activate RNase H, which cleaves the RNA moiety of a DNA–RNA heteroduplex and therefore leads to degradation of the target mRNA in the nucleus and cytoplasm. In addition, ASOs that do not induce RNase H cleavage can be used to inhibit translation by steric blockade of the ribosome in the cytoplasm [6]. When the ASOs are targeted to the 5′-terminus, binding and assembly of the translation machinery can be prevented [7]. Most mammalian RNAs undergo multiple post-transcriptional processing steps in the cell nucleus including addition of a 5′-cap structure, splicing and polyadenylation.
Regulation of RNA processing is another efficient mechanism in which ASOs can be utilized to regulate gene expression. Studies have been published documenting that ASOs can be used to destabilize pre-mRNA [8] and to regulate RNA splicing [9]. Another viable approach to reversibly ‘switch’ protein function is alternative splicing that generates RNA encoding antagonistic proteins (Figure 1). Whether RNaseH activity takes place favorably in the cytoplasm or in the nucleus is not well documented. However, most studies suggest that the most part of the inhibition takes place in the cytoplasm. Controversially, ASOs that targets pre-mRNA and are splice modulators are active in the nucleus.
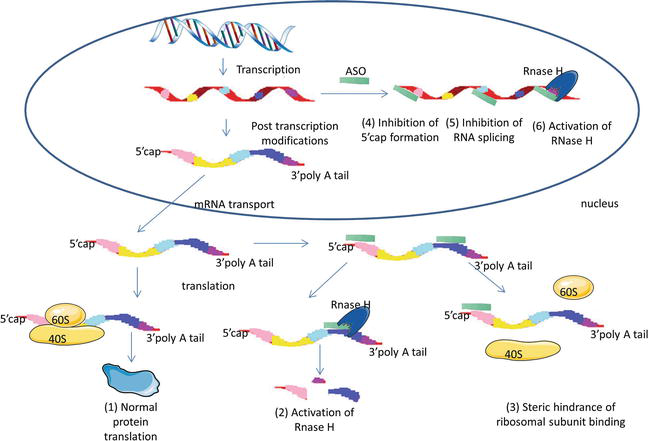
Figure 1.
ASOs mechanism of action. (1) In the absence of ASO, normal gene and protein expression is maintained. (2) Formation of ASO-mRNA heteroduplex in cytoplasm induces activation of RNase H, leading to mRNA degradation or (3) steric interference of ribosomal assembly. Alternatively, ASO can enter the nucleus and regulate mRNA maturation by (4) inhibition of 5′ cap formation, (5) inhibition of mRNA splicing and (6) activation of RNase H.
2.2. ASO’s design
There are several screening strategies to obtain potent ASO, such as using a computational algorithm in ASO design [10], mRNA walking [11], OD array [12] and RNase H mapping [13]. However, some of these approaches are labor intensive and require expensive automation equipment. Many factors can affect the strength and stability of the ASO-mRNA interaction, such as the mRNA secondary structure, thermodynamic stability and also the position of the hybridization site relative to functional motifs on the target RNA, such as the 5’ CAP region or translational start site.
To find a highly potent ASO, the “hit rate” can be increased by considering four parameters during ASO design:
2.2.1. Prediction of the secondary structure of the RNA
It is recognized that a secondary structure of the RNA accurately predicted, leads to effective ASO design [14, 15]. Some algorithm able to predict any single mRNA secondary structure and folding pattern are currently available, such as the mfold and the sfold program.
2.2.2. Identification of preferable RNA secondary local structures
In order for the ASO to be effective, it should target mRNA regions accessible to hybridization [16], such as joint sequences, internal loops, and hairpins of 10 or more consecutive nucleotides, usually located at the terminal end of the sequence [17]. It has been shown that highly conserved motifs are a good target of a potent ASO, whereas ASO targeting variable local motifs may induce non-sequence specific effects [18]. Therefore, the best approach to increase the ‘hit rate’ of potent ASO design is to target these conserved motifs among several optimal mRNA predicted secondary structures.
2.2.3. Motifs searching and GC content calculation
Although RNase activity is stimulated by the formation of the ASO-mRNA heteroduplex, leading to the mRNA degradation, it has been shown that this activity occurs independently of the ASO sequence, but is rather strongly correlated to the GC content which is also known to affect thermodynamic stability.
The perfect content of GC is still controversial; it is described that a strong ASO effect is observed with a percentage of 45–65% of G or C residues [10]; however, many of the ASOs used in therapy today do not have a GC content in this range.
2.2.4. Binding energy prediction
Thermodynamic energy is also important to a successful ASO design. Some available software can calculate thermodynamic properties between the target mRNA sequence and the ASO. To design a potent ASO, the binding energy between the ASO and mRNA should be DG37 ≥ −8 kcal/mol, whereas the energy for binding between ASOs should be DG37 ≥ −1.1 kcal/mol [19].
2.3. ASO’s chemical modification
The use of unmodified ASOs is limited as they have an overall charged property that prevents them from getting through the cell membrane and are rapidly attacked by all types of intracellular endonucleases and exonucleases, usually via 3′-5′ activity in biological fluid. In addition, the degradation products of phosphodiester ODs may be cytotoxic and also exert anti-proliferative effects. Moreover, some unspecific hybridization has been observed and finally for most applications immunostimulation has also been a matter of concern [20, 21]. This part will be further detailed in the toxicity chapter.
Numerous chemical modifications have been developed to improve nuclease resistance, extend tissue half-life, reduce non-sequence-specific toxicity, as well as to increase affinity and potency (Figure 2).
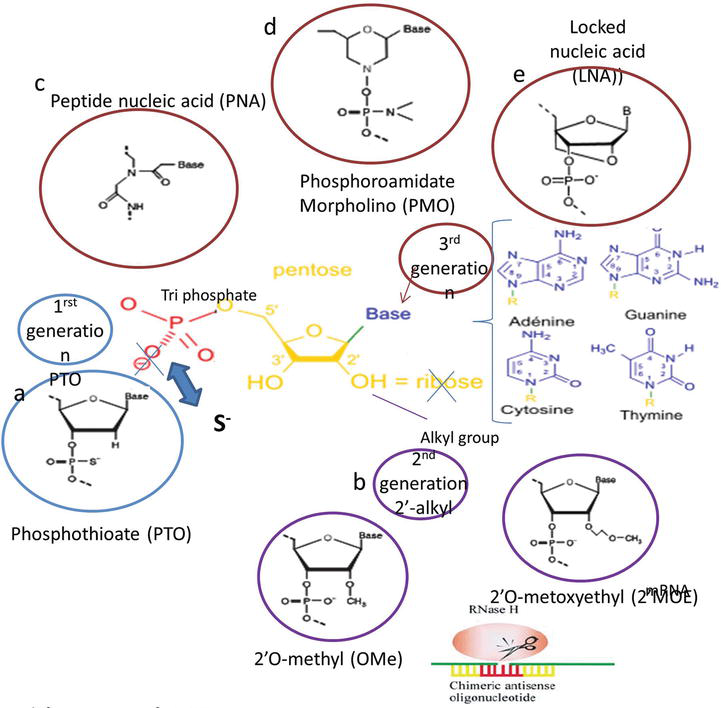
Figure 2.
Chemical modifications of ASO. (a) First-generation PTO, (b) second generation, RNase H cleavage induced by chimeric ASO, and (c–e) third-generation ASO.
2.3.1. First-generation ASO
First-generation ASOs are the ones having a phosphorothioate (PTO) modified backbone. These ASOs have a sulfur atom that substitutes the non-bridging oxygen atoms in the phosphodiester bond [22] (Figure 2a). PTO modification confers higher resistance to the ASO against nuclease degradation, leading to higher bioavailability. Additionally, they are highly soluble and have excellent antisense activity [23]. Finally, PTO-modified ASOs promote degradation of target mRNA by RNase H enzyme. Nevertheless, this modification may slightly reduce the affinity of the ASO for its mRNA target because the melting temperature (Tm) of the ASO-mRNA heteroduplex is decreased [20]. PTO modification is the most widely performed chemical modification of ASOs for loss-of-function studies in vitro and in vivo for gene target identification and validation. These data have led to the introduction of PTO ASOs into clinical therapeutic trials. Indeed, Fomivirsen, a currently FDA approved drug for clinical use, is a first-generation PTO-modified ASO [24]. Despite the fact the PTO modifications are the most widely used ODs, they have many properties which render them suboptimal antisense effector molecules. The PTO backbone is known to induce sequence-independent effects attributable to its length dependent high affinity for various cellular proteins. However, this seemingly negative property of PTO ASOs to interact with certain proteins proved to be advantageous for the pharmacokinetic profile. Their binding to plasma proteins protects them from filtration and is responsible for an increased serum half-life [25]. Finally, these oligonucleotides can interact with components of the innate immune system such as Toll-like receptors (TLRs), inducing an immune response and triggering cytokines expression and other genes coding for the nonspecific defense mechanisms [26].
Nevertheless, to try to solve the several non-specific problems [27], new chemical modifications have been developed. Modifications to the base, sugar and backbone have been identified that increase binding affinity for the target RNA.
2.3.2. Second-generation ASO
The second generation represents oligonucleotides in which the structural modification is not limited to the backbone linkage but additionally includes structural modifications of the ribose: ASO with 2’-O-alkyl modifications of the ribose were developed (Figure 2b). These modifications intend to improve binding affinity, to increase efficacy, to modulate the protein binding of oligonucleotides and to enhance nuclease resistance. 2’-O-Methyl (2’-OMe) and 2’-O-Methoxyethyl (2’-MOE) modifications of ASO-PTO are the most widely studied [28]. These second-generation ASOs are less toxic than PTO- modified ASOs and have a slightly enhanced affinity towards their complementary RNAs [29]. Moreover, an important aspect for these ASOs is that 2’modifications can reduce immunostimulatory effect [30]. However, 2’-OMe and 2’-MOE substitutions do not activate the RNase H to cleave the target mRNA, which decreases the efficacy of the ASO [31]. Indeed, mechanistic studies have been conducted to elucidate RNase H activity. They have shown that the flexibility of the ASO, the accessibility of the 2′-OH group of the RNA and the correct width of the minor groove of the ASO-RNA duplex are necessary for effective RNase H mediated mRNA degradation [32]. Since 2′-O-alkyl RNA ODs do not activate the RNase H, they inhibit mRNA expression only by a steric interference with translation. However, to increase its potency, an ASO should be able to induce cleavage by RNase H. Therefore, a chimeric ASO was developed (Figure 2b). It consists of a central ‘gap’ region containing 10 DNA or PTO DNA monomers and flanked on both sides (5’and 3’extremities) by approximatively 5 modified nucleotides such as 2′-OM or 2’-MOE (indicated by red and yellow regions of the OD in Figure 2b). This chimeric ‘gapmer’ ASO allows RNase H to sit in the central gap and to execute target-specific mRNA degradation; meanwhile, the flanking 2′-alkyl modified ends prevent nuclease cleavage of ASO. The 2′-O-MOE-PTO gapmers OGX-427 (Apatorsen) developed by Rocchi et al., directed against HSP27 and OGX-011 (Curtisen) against clusterin are currently undergoing clinical trials (Table 1). The ODs being tested clinically mainly incorporate relatively simple chemical modifications such as the PTO modifications and the 2′-alkyl modifications (first- and second-generation ODs).
2.3.3. Third-generation ASO
Third-generation ASOs have been developed to further improve nuclease resistance, increase binding affinity, and to enhance pharmacokinetics and biostability. They are characterized by chemical modifications of the nucleotide, and more precisely to its furanose ring [33]. Many modifications have been described, such as N3′-P5′ phosphoramidates, 2′-deoxy-2′-fluoro-β-D-arabino nucleic acid analogue (FANA), cyclohexene nucleic acids (CeNAs) and tricyclo-DNA (tcDNA), however, peptide nucleic acid (PNA), phosphoramidate morpholino oligomer (PMO) and locked nucleic acid (LNA) are the three most studied third-generation ASOs [34, 35].
It should be noted that there is no single modification that covers all the desired properties for a modified ASO. As described before, chemical modifications can improve ASO-RNA hybridization affinity, enhance nuclease resistance, decrease toxicity and modulate pharmacokinetics. An optimal antisense can be designed depending on its use by mixing and matching the numerous chemical modifications accordingly.
2.3.3.1. Peptide nucleic acid
PNA is a synthetic DNA mimic in which the deoxyribose phosphate backbone is replaced by polyamide linkages [36] (Figure 2c). PNAs are biologically very stable and have good hybridization properties. However, they do not activate mRNA cleavage via RNase activity, but rather block translation and thus protein expression, by forming sequence-specific duplex with mRNA, hence generating steric hindrance. Due to their neutral backbone, PNAs have low solubility and their cellular uptake remains a challenge for exploiting them as therapeutics antisense. PNAs delivery can be enhanced annealing a PNA strand with a negatively charged complementary oligonucleotide, and then enclosed the obtained duplex with a cationic lipid. To this end, PNAs are also conjugated to peptides for improving their cellular uptake [37, 38]. Furthermore, PNA can elicit antigen effects by hybridizing with double-stranded DNA [36, 39] resulting in transcriptional arrest. Substantial data have revealed the effectiveness of PNA in gene silencing in various in vitro models [39, 40].
2.3.3.2. Phosphoramidate morpholino oligomer
PMOs are non-charged ASOs whose pentose sugar is substituted by a morpholino ring and the inter-nucleotide linkages are phosphoramidate bonds in place of phosphodiester bonds [41] (Figure 2d). PMOs avoid the RNase H recruitment; their effect is primarily mediated by steric interference of ribosomal assembly resulting in translational arrest. Synthetic PMOs are resistant to nucleases degradation in biological fluid. Because their backbone is uncharged, PMOs are unlikely to form unwanted interactions with nucleic acid-binding proteins [42]. PMOs do not enter mammalian cells easily in culture, but it has been shown that conjugating it with peptides such as arginine-rich peptide (ARP) can enhance its cellular uptake and antisense potency [43]. Antisense PMO oligonucleotide, have shown efficacy in animal models in vivo and in human clinical trials [44, 45]. Indeed, a PMO antisense agent is currently in phase II clinical trials for restenosis, cancer and polycystic kidney disease.
2.3.3.3. Locked nucleic acid
Locked nucleic acids oligonucleotides figure among the most promising candidates developed the last few years. LNAs are chemically modified nucleotides with a ribose containing a methylene bridge between the 2′-oxygen and the 4′-carbon of the ribose [46]. LNA modifications improve significantly the ASO hybridization affinity towards mRNA target, due to the important increase in the thermal stability of the DNA/RNA heteroduplexes [47]. In addition, LNAs avoid nuclease degradation. As their ribose 2’-O position are modified, LNAs are not recruiting RNase H [48]. Nevertheless LNA nucleotides can be freely incorporated at the ends of RNA and DNA sequences to form chimeric oligonucleotides resulting in restoration of RNase H-mediated cleavage of mRNA. It has been shown that the chimeric LNA/DNA/LNA gapmer with seven to 10 phosphorothioate-modified DNA central gaps flanked by three to four LNA nucleotides on both 5′-end and 3’ends induces efficient mRNA cleavage, because of their high target affinity and nuclease resistance [29]. In the first in vivo study reported, the LNA ASOs appeared to be nontoxic in the optimal dosage. Therefore, full LNA and gapmers LNA·DNA·LNA ASOs seem to offer an attractive set of properties, such as potent biological activity and apparent lack of acute toxicity, making it a promising antisense agents [49, 50, 51]. Table 2 recapitulates the chemical modifications characteristics.

Table 2.
Chemical modification characteristics.
Despite the large number of molecules being evaluated in the clinical trials, the clinical progress of ASOs had to face many challenges; indeed, poor pharmacokinetics [52], poor cell membrane permeation [53], and off-target effects [27]. Last decade, the development of oligonucleotide delivery through lipid or polymer systems has improved the cellular uptake and the pharmacokinetic behavior [54].
3. ASO’s pharmacokinetics, toxicity and delivery
3.1. ASO’s pharmacokinetics and toxicity
The pharmacokinetics of ASOs has been extensively studied; they rapidly distribute to all tissues in vivo [55]. First and second-generation PTO ODs are broadly distributed to all peripheral tissues. The highest concentrations of ODs are found in the liver, kidney, spleen, lymph nodes and bone marrow with no measurable distribution to the brain [52]. Moreover, they not only distribute to tissues but also accumulate within cells in tissues [56]. The mechanism(s) by which ODs accumulate within cells following parenteral administration is currently unknown [57]. However, ODs that do not contain a PTO linkage appear to be rapidly excreted in the urine [58]. This difference in tissue distribution appears to be due largely to interactions with plasma proteins.
In summary, pharmacokinetic studies of PTO ASO demonstrate that they are well absorbed from parenteral sites, distribute broadly to all peripheral tissues, do not cross the blood-brain barrier (BBB), and are eliminated primarily by slow metabolism. Altogether, their pharmacokinetic properties depend on chemistry rather than sequence.
Concerning ASO’s toxicity, there are two broad categories of potential toxicities for ASOs; toxicities due to exaggerated pharmacology and toxicities due to non-antisense effects of the ASO. The former category of side effects results from the ASO binding to off-target RNA, producing an undesirable effect. The potential for such toxicities can be further minimized by vigilant selection of the drug target and homology searches against human genomic databases. The second category of potential toxicities (non-antisense effects) has been documented at higher doses of ASOs. In clinical trials PTO ASOs have proven to be safer than originally anticipated [31, 34, 35]. ASOs drugs principally induce mild-to-moderate toxicities related to the administered dose. In vivo, the two mains mechanism that induce acute toxicity through ASOs administration are for the first one the activation of the transient complement cascade and for the second one the inhibition of the clotting cascade. The increase in the clotting times is defined by a transient concentration-dependent escalation of activated partial thromboplastin times (aPTT) [59]. Another frequently occurring sub chronic toxicity is immune stimulation, manifested as splenomegaly, lymphoid hyperplasia and diffused multi-organ mixed mononuclear cell infiltrates [60]. This is due to an unmethylated cytosine-phosphorous-guanine (CpG) motif in the ASO sequence that can be recognized by Toll-like receptor (TLR)-9 in immune cells. This results in the release of cytokines, interleukin (IL)-6, IL-12, interferon (INF)-γ), B cell proliferation, antibody production and activation of T lymphocyte and natural killer (NK) cells [61]. In order to avoid the immune response, the cytosine of the sequences including CpG motif are substituted by methylated cytosine. Also, for preventing this side-effect, the sequences of ASOs drug can be designed without CpG motif. These both stimulatory immune responses are related to the sequence and the chemical modification of the ASOs [6]. Indeed, second-generation ASOs have been shown to have smaller immune stimulation that ASOs PTO. In addition, introduction of LNA into the PTO-ASO has been shown to reduce, and even eliminate, CpG dinucleotide-mediated immunostimulation [62, 63]. When ASO’s plasma concentration is high, some toxicities have been observed, such as elevated liver enzyme such as aspartate aminotransferase (AST) and alanine amino-transferase (ALT), thrombocytopenia, and hyperglycemia [52].
3.2. ASO’s delivery
Although the clinical relevance of ASOs has been demonstrated, inefficient cellular uptake, both in vitro and in vivo, limit the efficacy of ASOs and has been a barrier to therapeutic development. Cellular uptake can be <2% of the dose resulting in too low ASO concentration at the active site for an effective and sustained outcome [22]. Indeed, many researches are conducted on the improvement of ASO delivery via the use of delivery vehicles.
3.2.1. Mechanism of cellular uptake
Cellular uptake refers to the combination of both OD membrane-binding and internalization. It is challenged by numerous barriers, most importantly, the lipophilic nature of the cell membrane, which makes the passage of these large anionic molecules complicated. Several barriers to cellular uptake exist such as the lipophilic cell membrane, through which these large, anionic molecules must pass to reach the site of action. In cultured cells, the internalization of naked ASOs is generally inefficient, with only a few ASO molecules actually penetrating the cell [21]. Exogenously administered ASOs enter cells in vitro by a combination of fluid-phase (pinocytosis), caveolae potocytosis, adsorptive and receptor-mediated endocytosis (Figure 3), cellular uptake mechanisms being different depending on the ASO chemical structure. However, this results in a trafficking problem because not all of the internalized ASO will be able to reach their target and interact with it. This is because the majority of internalized ASO is sequestered into endosome or lysosomal compartments [21]. A significant amount of the OD is also compartmentalized within other cellular organelles, such as the Golgi complex and the endoplasmic reticulum [64].
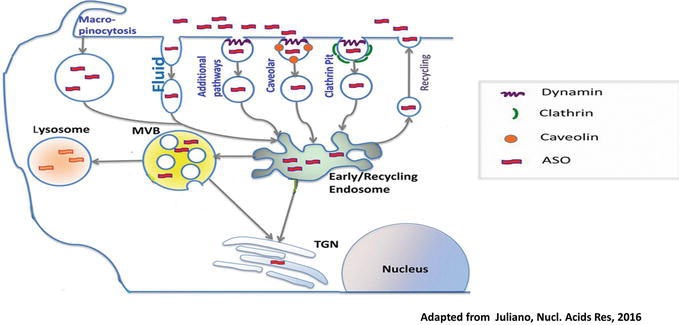
Figure 3.
Cellular uptake and intracellular trafficking of oligonucleotides. Oligonucleotides enter cells via several endocytotic pathways. Most of ODs are recycled in early recycling endosomes. ODs accumulate in endosomes, multivesicular bodies (MVB) and lysosomes. A significant amount is compartmentalized in cellular organelles like the trans Golgi network (TGN).
To improve cellular uptake and OD’s activity, a range of techniques and transporters have been developed [65, 66]. Simultaneously, the use of these vectors increases the stability of ODs against nuclease digestion and allows the use of lower concentrations of ODs.
3.2.2. General delivery methods
OD delivery can occur through carrier molecules. Receptor-mediated endocytosis-directed uptake utilizes import mechanisms already present in the cell membrane for the uptake of biomolecules necessary for cell function [67]. ODs can be linked directly to a carrier protein via a covalent bond or non-covalently via poly-L-lysine (PLL)-carrier conjugates. The choice of the carrier is dependent on its known ability to bind to specific cell membrane receptors and accumulate in the cell via endocytosis. Not only OD’s internalization can be potentially improved, but cell-specific delivery can also be achieved by targeting receptors exclusively expressed or over-expressed on certain target cells. However, the OD is generally sequestrated in endosomal compartments, thereby limiting the utility of this method for delivery [22]. The use of peptides can help overcome this issue. Indeed, as peptides have membrane translocation properties, they can increase ODs passage through the plasma membrane. For example, fusogenic peptides have been used to promote peptide fusion of OD-peptide conjugates with either cell or lysosomal membranes [68]. To facilitate OD transport to the nucleus, nuclear localization signals (NLS) can be used for ODs that inhibit pre mRNA splicing [69]. Another peptide used is CPP, a short peptide sequence (<30 amino acids) with net positive charge that uses an energy dependent pathway to allow internalization of large molecules like ASOs. Commonly used CPP include penetratin, HIV TAT peptide and transportan. Through disulfide bond, ASOs and CPP can be conjugated. To evaluate ASO-CPP effect on mRNA degradation in vitro and in vivo, PNA is the most used [43, 70, 71]. An interesting variation is to couple two different ASOs to a single CPP thus addressing two targets simultaneously [72]. The most dramatic advance in OD targeting has involved delivery via the asialoglycoprotein receptors (ASGR). ASGPR, also known as the Ashwell receptor, is a lectin that is abundantly expressed on hepatocytes [73] and clears serum glycoproteins by receptor-mediated endocytosis [74]. The functional receptor is a trimer comprised of two proteins and exhibits high affinity for N-acetyl galactosamine (GalNAc) terminated oligosaccharides [75]. A major breakthrough came from researchers who developed multivalent GalNac conjugated siRNAs that bind to the ASGR [76]. The conjugates were effectively taken up into primary mouse hepatocytes by a receptor-specific mechanism, leading to silencing of targeted genes. GalNac based conjugates have similarly been used to transport ASOs to the liver in mice with good effects on reduction of target gene expression [77]. Indeed, GalNac-ASOs showed high affinity for mouse ASGPR, which resulted in enhanced ASO delivery to hepatocytes. Furthermore, once inside the cell, GalNac-ASO’s metabolism leads to ASO release in the liver, thus acting like a prodrug targeting hepatocytes. Moreover it improved potency and extent the effect of both antisense targeting human transthyretin (TTR) and human apolipoprotein C-III in transgenic mice. This study highlighted several of the virtues of conjugates including use of a molecularly defined entity, high tissue and cell selectivity and lack of substantial toxicity. The successes with glycoconjugates in the laboratory have facilitated their rapid translation to clinical evaluation, with several hepatic genes being addressed. Finally, an additional approach to OD internalization is to generate transient permeabilization of the plasma membrane and allow naked ODs to penetrate into the cells by diffusion. It consists of inducing transitory pores in the membrane, either chemically by streptolysin-O-permeabilization, mechanically by microinjection, or by electroporation [78] or ultrasound.
All of these methods, under defined circumstances, can permit charged or uncharged ODs to enter cells rapidly and localize in the nucleus, where they produce antisense inhibition of gene function.
Nevertheless, one of the most promising strategies used to overcome cellular barriers is the use of nanotechnology. Indeed, nanocarriers offer the possibility to encapsulate drugs but also nucleic acid, to protect it from degradation, enhance its distribution to the tissues and the cells, but also reduce toxicity and thus secondary effects by targeting specific organs. It is a very promising tool for combination therapy and theranostics applications. To bypass the limitations of ASO-based therapy, like insufficient stability and low intracellular delivery and distribution, Dr. Rocchi’s lab recently developed a modified first-generation ASO by using a lipid-conjugated oligonucleotide modification (LASO) able to overcome the problems to ASO administration. The idea is to improve ASO’s stability, efficiency and biodisponibility by adding a hydrophobic lipid chain at the 5’end of the hydrophilic DNA sequence. As amphiphiles, LASOs self-assemble in aqueous media with an encapsulation rate of 100%, which yields in small spherical objects of ~11 nm in diameter [79]. Due to their small size, these nanomicelles, can accumulate inside the tumor via EPR effect (enhanced permeability and retention effect), which permits passive targeting and reduced systemic toxicity. Interestingly, transfection with these lipid-modified ASO leads to rapid and prolonged internalization via micropinocytosis with no transfecting agent. Interestingly, the addition of the lipid, does not affect LASO’s efficacy in vitro and in vivowith little or no toxicity in animal models.
4. Discussion/conclusion
Antisense oligonucleotide based therapies are promising. They can be used for human therapy and can specifically inhibit target genes. Despite the promising progress since their discovery, only a few have been approved for clinical use. This low success rate can be explained by their anionic charge which prevents them from getting through the cell membrane and are rapidly destroyed by nuclease. To address these problems many chemical modifications have been developed. Finally, improvement of their delivery to the tissues, and thus their efficacy is an important issue that has to be addressed. To solve this problem, many strategies have been studied; nanotechnology is one of the most promising to overcome the cellular barriers for the ASO delivery towards targeted therapy.
Source: intechopen.com



